
Scalable Molecular Dynamics with Agentic AI

Molecular Dynamics (MD) is the workhorse of atomistic simulation. Axon is equipped to launch and run MD at production scale, using familiar MD engines, and on state-of-the-art accelerating hardware.
Challenges in Molecular Dynamics
Molecular dynamics (MD) uses equations of motion to simulate the movement of atoms and molecules over time, enabling researchers to observe how molecular systems evolve. These simulations allow for insight into binding interactions, conformational dynamics, mechanical properties, etc. with applications in drug discovery, materials science, chemistry, and more.
While a powerful observational tool, MD has long been bottlenecked by time/lengthscale and sampling limitations: timesteps need to be very small (~1 fs) for atomistic simulations to be meaningful, so even simulations that run for hours to days or weeks on dedicated hardware yield enormous amounts of data, but only microseconds of simulation. Similarly, the larger the system, the more compute that is required. As a result, it’s often intractable to explore the full energy landscape of molecular systems.
The utility of MD also depends on simulation accuracy, which is reliant on the energies and forces computed between atoms. These computations are performed with forcefields that parameterize interactions between different types of atoms, or more recently, with machine-learned interatomic potentials (MLIPs) trained on quantum data. Various forcefields, and the MD engines equipped to run with them, have significant tradeoffs in speed, accuracy, and domain coverage, meaning that simulating different molecular systems can require drastically different setups. Furthermore, these different software ecosystems have varied input formats and interaction patterns, including hardware (GPU) communications, so translating between different packages can be extremely complex.
Axon in Action
Axon harnesses AI to address many of these challenges. Agents can be taught the languages of different software ecosystems and to translate between them so that researchers are not bottlenecked by syntax. And by handling compute allocation, parallelization, and workflow management, Axon enables researchers to be optimally efficient with time and resource costs, as well as to run multiple experiments simultaneously to get the results they need faster.
To demonstrate this in action, we collected timing data on Axon deployed to simulate a few pharma-relevant proteins from PDB using 3 different MD engines and several hardware (GPU) configurations. Notably, we chose a single forcefield that was appropriate for protein simulations and was compatible with all 3 MD engines, but Axon can access other well-known forcefields and MLIPs.
Configuration details:
Forcefield: AMBER99SB-ILDN with TIP3P water (unified via GROMACS master topology)
Simulation targets: DHFR (1DRF; ~3K atoms), Lysozyme (1AKI; ~2K atoms), ApoA1 (1AV1; ~13K atoms), ADH (2OHX; ~11K atoms), GluTS (1FPY; ~90K atoms)
GPU types: T4, L4, A10G, L40S, A100-40GB, A100-80GB, H100, H200
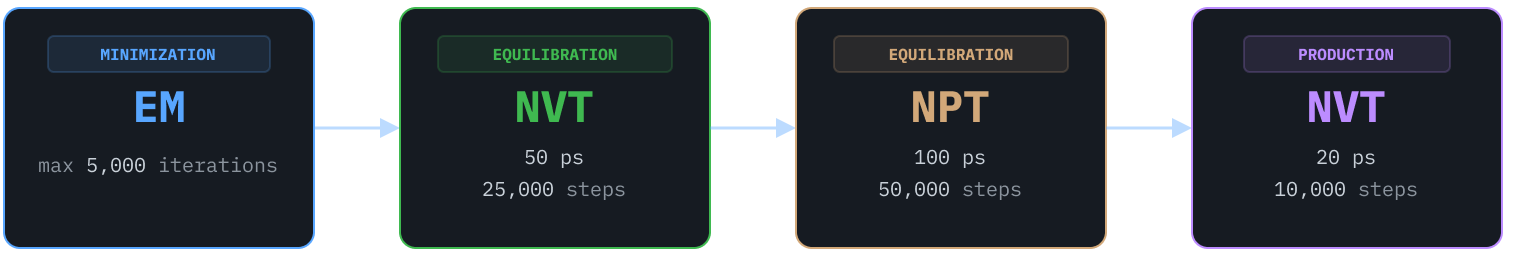
With straightforward prompting, Axon performed necessary preparatory work including structure cleaning (with PDBFixer), parameterization (GROMACS), and solvating the simulation boxes. Axon also created the input files for each MD engine, including structure topology and data and parameter files. Finally, Axon ran a typical simulation workflow using each MD engine as follows, using a 2 fs timestep and default thermostats for each engine:
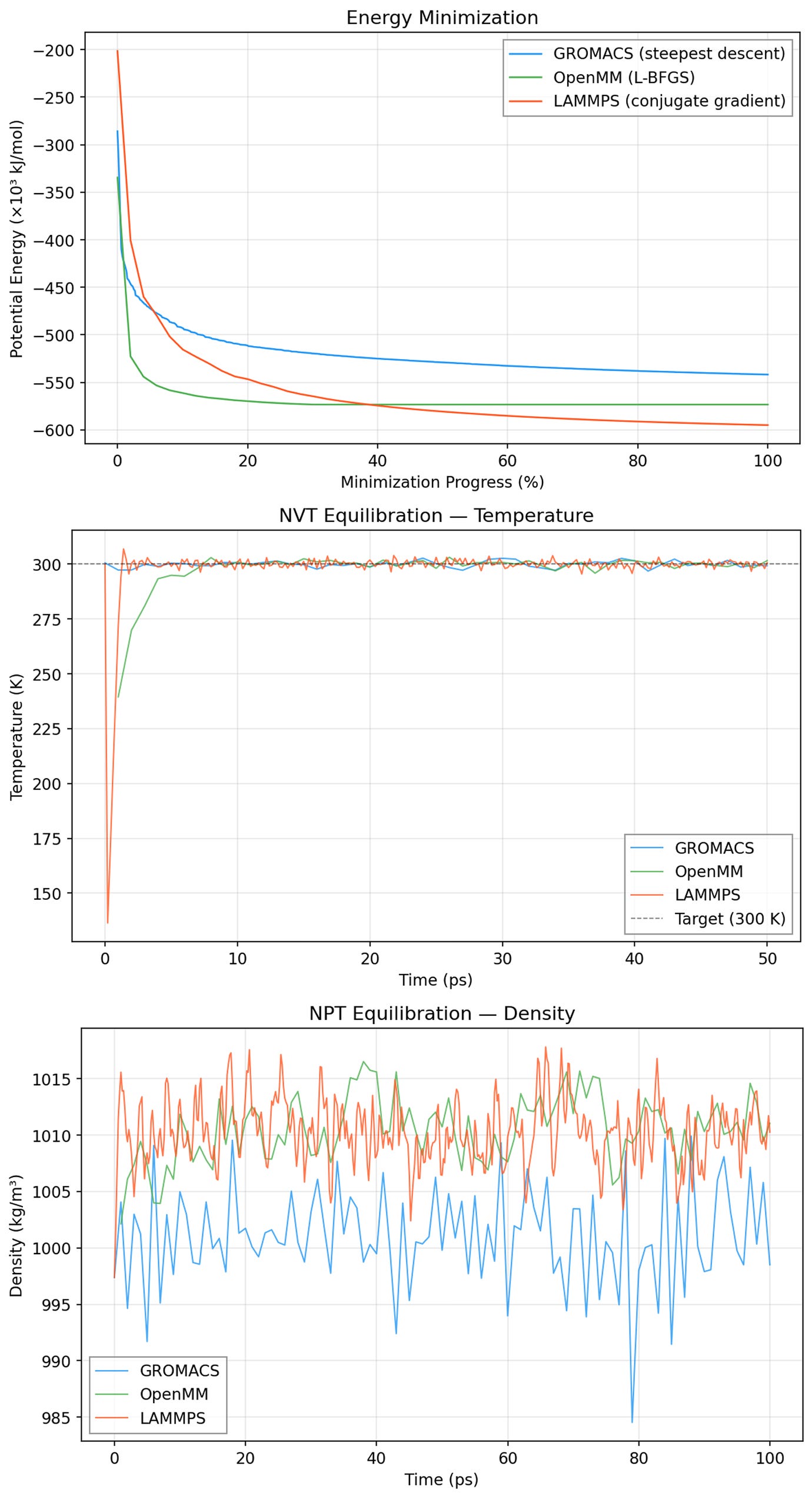
We inspected results from energy minimization and equilibration stages by prompting Axon to collect and visualize relevant data. In the example below, we observe the potential energy of the lysozyme (1AKI) system through the energy minimization step, and it is evident that potential energy decreased steadily and plateaued at similar values across all three MD engines. Similarly, we see temperature stabilize right around 300K in NVT equilibration (center), and relatively stable box density in NPT equilibration (bottom). Here, the GROMACS density is likely a little lower than that in OpenMM or LAMMPS because of its different handling (potential-shift vs. plain cutoff) of Lennard-Jones truncation, but the ~0.01% shift should not meaningfully effect typical MD outcomes. These results give us confidence that energy minimization and equilibration proceeded correctly with all three engines.
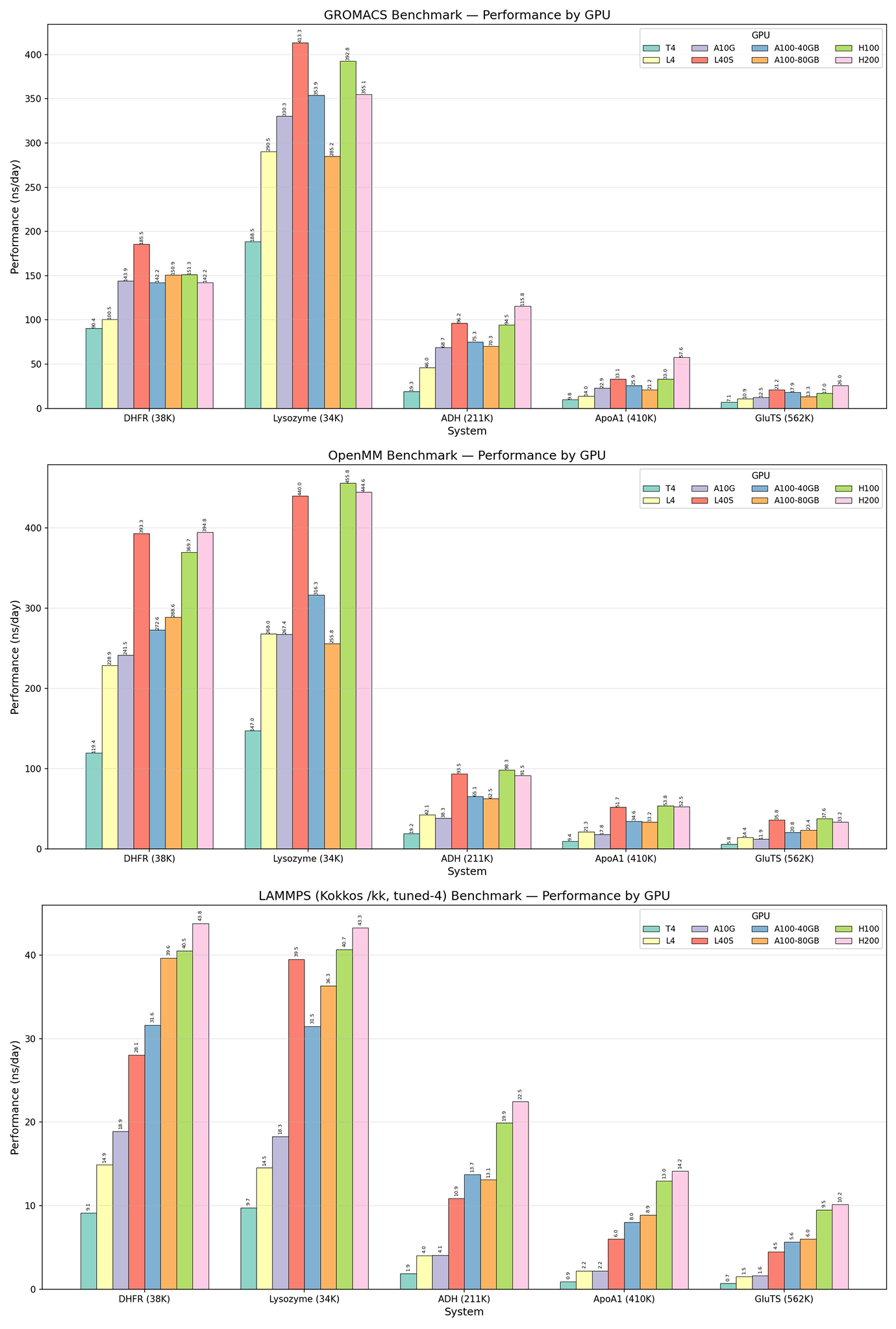
With the relaxed structures from each MD engine, we ran “production” MD of each protein/engine configuration on 8 different GPUs, simulating the proteins (with no external forces) just long enough to collect robust timing data. As expected, the newest and most powerful GPUs (L40S, H100, H200) generally showed higher simulation throughput than mid-tier (A100, A10G) and entry-level (L4, T4) GPUs. Similarly, the smaller protein systems showed higher simulation throughput than larger systems, which are more computationally demanding.
Interestingly, GROMACS and OpenMM showed relatively similar simulation throughput across protein systems and GPUs while LAMMPS was around an order of magnitude slower. GROMACS and OpenMM are more commonly used for protein simulations and are more optimized for biomolecular tools and force fields out-of-the-box. LAMMPS, meanwhile, is extremely flexible and excels at materials science simulations and coarse-grained models, so is less specialized to proteins. Some brief searching indicated that building LAMMPS with mixed precision would be a significant accelerator—something for us to try next time! Here, we primarily wanted to show the capacity to run various configurations of software and hardware, allowing researchers to compare performance and costs (available but not shown here) and to choose the tools that work best for their contexts.
A New Paradigm for MD Research
Running very short simulations of well-studied proteins is not particularly interesting from a scientific standpoint (although it does still enable Axon to generate visually striking animations like the one of lysozyme up top!). What we really want to emphasize here, however, is that we were able to simulate 5 proteins with 3 different MD engines and to collect timing data across 8 different GPUs—a total of 120 different configurations—all run in parallel through a few simple prompts. If done manually, setting up and translating even one system between different MD engines is often a massive headache, not to mention the infeasibility of accessing and managing 120 independent GPUs and their data.
Instead, Axon manages file handling and serves as a collaborator and assistant already well-versed in various computational chemistry methods and tools. Axon is also fully integrated with AI infrastructure that launches and scales containers in moments, and enables elastic GPU scaling at competitive market rates, so Axon’s tasks can be scaled arbitrarily and the user only ever pays for the compute that is actively used. All this means that, for the first time, researchers can explore many different software and hardware configurations all within the same platform, creating a new mode of research that can accelerate exploration and optimization with ease.
We’ve demonstrated Axon’s elasticity and scalability on a few molecular dynamics tasks here. We know this is the tip of the iceberg and would love to connect with researchers in MD or related fields—we’re keen to learn from YOU what packages, tools, and work patterns would bring the most utility. We invite you to get in touch and try out Axon on your own research!
We're super excited about Axon and have way more to say about it than fits in a blog post. If you have questions or comments or just want to talk with us, shoot me a message at sabrina@mirrorphysics.com.
I appreciate the effort. I used to run several calculations with different software packages, and I can see that saving time if that integrates well with a particular wokflow. But I cannot wrap my head around why one would necessarily need AI for that.. Maybe if there is a new software update and things change, Mirror would pick it up and change things appropriately? Or flag a bad initial guess state due to "bad" initial geometry?
Other questions: What does "With straightforward prompting" look like? A live/recorded demo would be really cool. Can Mirror build the workflow for you with just prompting, yielding something like a make.com or n8n scenario? Do the nodes/touchpoints need a human in the loop? I imagine me setting it up for the weekend just to see on Monday that the jobs and thus the workflow failed miserably due to, say, convergence issues. :)